
2020-05-11 19:10:00

在一致性评价的大背景下,国家局根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)以及《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号),以及顺应药物研究开发的现实出发,参考了FDA.Guidance for industry: “Waiver of in invo bioequivalence studies forimmediate-release solid oral dosage forms based on a BiopharmaceuticsClassification System”等指南,制定了我国的人体生物等效性试验豁免指导原则。该指导原则是基于国际公认的生物药剂学分类系统(BiopharmaceuticsClassification System,以下简称BCS)起草,可用于仿制药质量和疗效一致性评价中口服固体常释制剂申请生物等效性(Bioequivalence)豁免。BCS系统是按照药物的水溶性和肠道渗透性对其进行分类的一个科学架构。当涉及到口服固体常释制剂中活性药物成分(Active Pharmaceutical Ingredient,以下简称API)在体内吸收速度和程度时,BCS系统主要考虑以下三个关键因素,即:药物溶解性(Solubility)、肠道渗透性(Intestinal permeability)和制剂溶出度(Dissolution)。
根据BCS分类系统,药品被分为以下四类:
第一类:高溶解性、高渗透性(High Solubility- HighPermeability)
第二类:低溶解性、高渗透性(Low Solubility-HighPermeability)
第三类:高溶解性、低渗透性(High Solubility-LowPermeability)
第四类:低溶解性、低渗透性(Low Solubility-LowPermeability)
指南指出:当口服固体常释制剂在体内的溶出相对于胃排空时间快或非常快,并且具有很高的溶解度时,药物的吸收速率和吸收程度就不会依赖于药物的溶出时间或在胃肠道的通过时间。因此,在这种情况下,对于BCS分类1类和3类的药物,只要处方中的其他辅料成分不显著影响API的吸收,则不必证明该药物在体内生物利用度和生物等效的可能性,即生物等效性豁免。
(一)对于BCS1类的药物需要证明以下几点:
1.药物具有高溶解性;
2.药物具有高渗透性;
3.仿制和参比制剂均为快速溶出(30min>85%),并且制剂中不含有影响主药成分吸收速率和吸收程度的任何辅料。
(二)对于BCS3类的药物需要证明以下几点:
1.药物具有高溶解性;
2.仿制和参比制剂均具有非常快速的溶出(15min>85%));
3.仿制制剂和参比制剂应处方完全相同,各组成用量相似。
药物溶解性的测定数据相对简单易得,且具有良好的重现性。原则中对高溶解性定义为:单次给药的最高剂量对应的API在体积为250ml(或更少)、pH值在1.0—6.8范围内的水溶性介质中能完全溶解。快速或非常快速溶出也可以采用溶出仪进行测定获得。
然而,想要BE豁免BCS1、3类药物,存在两大阻碍:
(1)药物渗透性;(2)辅料不影响药物吸收速度和吸收程度,这二者均不易证明。因仅仅通过文献报道的研究来说服审评机构是及其困难的,这也能从FDA、PMDA等监管机构对BE豁免所持的谨慎态度上可见一般。对于可能影响药物吸收转运的辅料包括表面活性剂、渗透压调节剂(甘露醇、山梨醇等)、以及脂质(硬脂酸甘油酯、油酸等),如参比制剂处方包含了此类物质,监管机构对于BE豁免上至少应会要求仿制药企业证明其用量完全一致,甚至可能还要求研究表面活性剂分子量分布或者表面活性剂溶出速率,均不易证明。
对于渗透性,目前FDA、EMA等审评综述中都会披露原研企业所做的药物BCS分类研究结果,但其可信程度需要仿制药企业进行深入而细致地分析与核实,以免掉进“坑”中而不自知。以达沙替尼为例,在FDA公开的综述文件Sprycel_ClinPharmR中,我们可以找到其BCS为2类,即低溶解度高渗透性。原文可在综述中第39页找到,如下:

溶解度数据也在同一页内,溶解度为pH依赖性,pH7.0时<1μg/ml,pH6时~8μg/ml,如下:

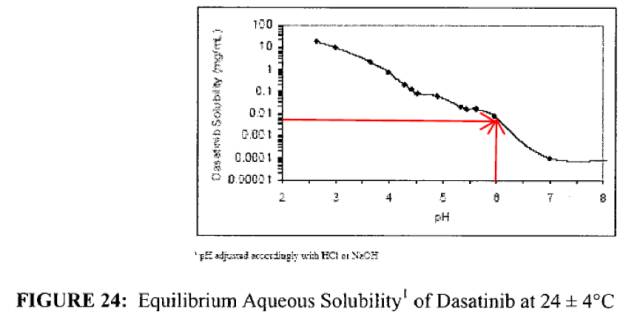
渗透性研究结论如下:

资料显示采用了caco-2细胞实验模型(一种人克隆结肠腺癌细胞,结构和功能类似于分化的小肠上皮细胞,具有微绒毛等结构,并含有与小肠刷状缘上皮相关的酶系,可用来模拟体内肠转运的实验。)通过与FDA推荐的高渗透性内标药物美托洛尔和地塞米松比较,确定达沙替尼为高渗透性药物。如就此认为其定义为高渗透性是合理的,那我们就会掉入 “陷阱”。
首先,FDA审评员在上图中指出渗透性结果应refer to section 2.2.5.5 for in vivo permeability。我们找到p26中此部分内容,其旨在研究达沙替尼是主要通过肾脏或肝脏代谢的消除途径;通过测定放射性标记药物,表明达沙替尼肾脏消除量非常低,而超过85%的通过粪便消除,粪便中原形药比例为19.1%,结果如下图:
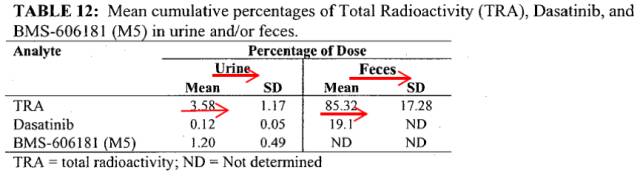
同时给药后2h血浆中达沙替尼M21代谢物(sulfate conjugate,磺酸复合物)水平达到总放射活性的9.5%,表明达沙替尼可能会在胃肠道经硫酸脂酶代谢。

综上分析,达沙替尼原形药19.1%通过粪便排泄,表明其体内绝对生物利用度应小于85%,且药物在胃肠道存在被代谢可能。
FDA 2015年BE豁免指南[1]中对于高渗透性的定义为:
A drug substance is considered to be highly permeable when the extentof absorption in humans is determined tobe 85 percent or more of an administered dose based on a mass balancedetermination (along with evidence showing stability of the drug in the GItract) or in comparison to an intravenous reference dose.即相对与所给药剂量,口服吸收程度≥85%(基于质量平衡研究-同时证明药物在胃肠道的稳定性;或与静脉注射相比)。
高渗透性测定依据:
Thepermeability class of a drug substance can be determined in human subjects using mass balance, orabsolute BA, which are the preferred methods, or intestinal perfusionapproaches. In many cases, a single method may be sufficient: (i) when theabsolute BA is 85 percent or more, or (ii) when 85 percent or more of theadministered drug is excreted unchanged in urine, or (iii) when 85 percent ormore of the administered drug is recovered in urine as parent and metaboliteswith evidence indicating stability in the GI tract. 渗透性可通过人体受试者的质量平衡或绝对生物利用度(优先选择)或肠道灌注研究确定;如下任一方法均可:1)绝对生物利用度≥85%;2)或≥85%药物以原型通过尿排泄;3)或≥85%药物以原型和代谢物形式从尿液回收-同时证明药物在胃肠道的稳定性(胃肠道稳定的前提下尿液中代谢物为系统代谢而非胃肠道代谢后再吸收排泄);而药物在USP推荐的模拟胃液1h和模拟肠液中3h的降解>5%被认为不稳定。
因此,可以认为原研对达沙替尼体内质量平衡研究结果不支持高渗透性判断依据:85%以上的原型药物从尿液中排出且胃肠道稳定,更没有绝对生物利用度>85%数据,故参考原研审评综述资料所公布的BCS分类结论,就很可能会误导仿制药研究人员。
其次,我们如能进一步仔细阅读,还可在p35页发现药物的外排蛋白(p-gp)底物研究,如下图:

研究发现在Caco-2试验中药物浓度为50μM时,渗透系数A-B端为102nm/s,B-A端为222nm/s,表明达沙替尼为p-gp底物。这份研究结果中可以看到FDA对其研究提出要求:应采用一系列药物浓度而不仅仅是单一的50μM,以及使用已经p-gp底物进行阳性对照。我们应该看到,达沙替尼分子量488,50μM 浓度换算得24.4μg/ml,该浓度已经远远超过药物在pH7介质中的饱和溶解度(<1μg/ml),意味着其渗透性结果和p-gp结果都不能反应体内真实情况。这是因为在采用caco-2细胞模型研究渗透性时,药物通常使用DMSO溶解配成高浓度溶液,再稀释到细胞培养基中,才能获得50μM的浓度;而人体内因为药物溶解度低,几乎不可能达到该浓度,其结果就会偏离实际,产生误导的结论。药物真实渗透性需要考虑药物从A-B端的渗透系数与p-gp等外排蛋白对药物的外排作用,如果高浓度的药物可以导致p-gp饱和,则会提高其渗透性,但未饱和p-gp时的渗透性会降低。对达沙替尼而言,其为p-gp底物,且肠道饱和溶解度明显低于50μM,所测定得到的A-B端渗透系数就可能是偏高的;这也是FDA要求研究不同药物浓度下的渗透系数之原因。
药物主要在小肠吸收,小肠不同区域的pH值不同,从偏酸性到中性直至弱碱性,也会影响到酸碱药物的解离程度从而影响药物吸收;因此,caco-2细胞试验中A端(细胞顶膜)的pH也应考虑在内。研究发现,布洛芬、酮洛芬、匹罗昔康等酸性药物在pH5.0的渗透系数显著高于pH7.4;而美托洛尔、噻吩洛尔等碱性药物在pH7.4的渗透性显著高于pH5.0;而这些药物在人体几乎完全吸收,生物利用度>90%;因此,caco-2细胞试验研究渗透性时,培养基的pH值应当考虑在内的。caco-2细胞实验适合用于被动转运的药物;而对于具有主动转运机制的药物而言,caco-2细胞实验获得的低渗透性结论就会导致假阴性判断。如头孢氨苄为二肽转运体底物,尽管其caco-2渗透性低,其体内吸收完全。同时,对难溶性药物溶解使用的DMSO在培养基中超过1%时,就会影响caco-2细胞膜的紧密连接,使其出现假阳性结论。其他影响细胞紧密连接致密性的还包括药物浓度、细胞培养条件等,不同实验室之间的结果往往难以重现。
总之,仿制药开发过程中,监管机构对于BE豁免的审批还是很谨慎的。熟悉FDA仿制药申报的专家曾透露,在美国要证明一个药物的高渗透性所需的工作量和费用也不亚于一个BE,故美国很少普通口服制剂的仿制药真正通过BE豁免上市。我国的普通固体口服制剂如希望BE豁免,要说服CFDA也同样不易。
我们珍惜您每一次在线咨询,有问必答,用专业的态度,贴心的服务。
让您真正感受到我们的与众不同!

为了提供更好的合作体验,我们提供专业的专线咨询和上门洽谈服务

关于产品的使用、售后服务等各种常见问题

从销售支持到后期维护,我们提供全方位360°的立体化服务,只为您的至臻追求
 全国销售服务热线:400-100-1649
全国销售服务热线:400-100-1649 
